
 Discussion
Discussion
Stages of Huntington’s disease
Last updated Aug. 20, 2025, by Marisa Wexler, MS
 Fact-checked by Marta Figueiredo, PhD
Fact-checked by Marta Figueiredo, PhD
Huntington’s disease is a genetic and progressive neurodegenerative disorder marked by motor, cognitive, and behavioral symptoms that gradually worsen over time.
Huntington’s disease progression is typically evaluated using staging systems that describe Huntington’s disease stages, from early stages where symptoms are mild and patients are mostly independent to later stages where symptoms become very debilitating and patients need regular assistance.
While there is no Huntington’s disease treatment that can slow or stop disease progression, learning about the different stages of Huntington’s disease can provide clarity on what to expect and help with long-term planning.
What are the 5 stages of Huntington’s disease?
Several different systems are used to describe Huntington’s disease stages. Sometimes, people will talk broadly about early, middle, and late stage Huntington’s, but some staging systems also include the very earliest stages of Huntington’s, prior to symptom onset.
Pre-manifest or preclinical Huntington’s disease broadly refers to the time before Huntington’s disease symptoms first appear. Increasing evidence shows that people carrying a Huntington’s-causing mutation may experience subtle cognitive and behavioral changes more than a decade before the onset of hallmark motor symptoms. This is called a prodromal stage.
But even before these subtle symptoms, certain Huntington’s markers, including changes in brain scans, may be detectable and relevant for eligibility in clinical trials of experimental treatments that aim to slow or stop symptom onset.
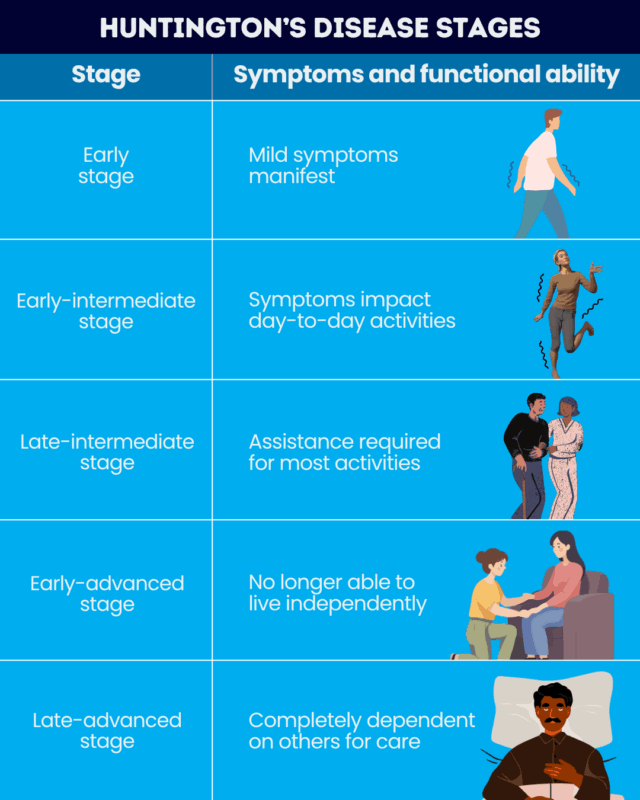
A five-stage system is typically used to describe the disease’s progression after symptom onset. This system is based on a validated test, called the Total Functional Capacity (TFC), that is a part of the Unified Huntington’s Disease Rating Scale.
TFC evaluates parameters including capacity for work, finances, domestic chores, daily life activities, and level of care. Total scores, which reflect a person’s functional status, range from zero to 13, with higher scores indicating better function. The five Huntington’s stages include:
- early stage (stage 1), with TFC scores ranging between 11-13
- early-intermediate stage (stage 2), with TFC scores of 7-10
- late-intermediate stage (stage 3), with TFC scores ranging between 3-6
- early-advanced stage (stage 4), with TFC scores of 1-2
- late-advanced stage (stage 5), with a TFC score of zero
It’s important to note that Huntington’s disease progression may look different for every person with the disease, depending on factors ranging from underlying genetics to lifestyle habits.
In addition, these stages refer mainly to adult-onset Huntington’s – juvenile Huntington’s disease, where symptoms manifest before 20 years of age, tends to progress faster, and symptoms may manifest differently.
Early-stage Huntington’s
Early stage Huntington’s disease starts when hallmark motor symptoms that allow an official diagnosis to be made first appear. This typically happens between the ages of 35 to 44.
In this stage, which is thought to last up to eight years, patients typically experience mild symptoms and are generally able to carry out most day-to-day activities independently. Still, people with early-stage Huntington’s may already benefit from creating plans and systems for the future.
Symptoms in the early stages of Huntington’s disease may include:
- chorea (uncontrollable muscle movements) in the arms, legs, or face
- coordination problems that can result in clumsiness and stumbling
- difficulty concentrating or thinking through complex problems
- mood changes like depression or irritability
Early-intermediate stage Huntington’s
As Huntington’s progresses into the early-intermediate stage, which is thought to occur three to 13 years after disease onset, symptoms become more obvious and burdensome.
People in this stage require more assistance and may no longer be able to work, but can still handle some things independently, like domestic chores, daily activities, and finances.
Symptoms in this middle stage Huntington’s may be similar to but more pronounced than those of early-stage Huntington’s, while new symptoms may also appear. Symptoms include:
- more substantial chorea and issues with balance, which increase the risk of falling
- loss of ability to work or drive
- increased difficulty organizing, planning, remembering, and processing information
- more pronounced mood and psychiatric changes, including irritability, anxiety, impulsiveness, and delusions or hallucinations
- trouble swallowing
- difficulty speaking intelligibly
Late-intermediate stage Huntington’s
The period comprising five to 16 years after disease onset is typically classified as late-intermediate stage Huntington’s. People in this stage need major help with most day-to-day activities, domestic responsibilities, and financial affairs.
Symptoms of late-intermediate stage Huntington’s will typically include all of those in the early-intermediate stage, but these will be even more severe and debilitating.
Early-advanced stage Huntington’s
People entering the early-advanced stage of Huntington’s, which generally occurs nine to 21 years after disease onset, start to need substantial assistance in essentially all areas of day-to-day functioning.
In these later stages of Huntington’s disease, people may experience
- severe chorea, although this symptom typically eases in later stages
- worsening of other motor problems, including rigidity, slowness of movement, involuntary muscle contractions that lead to abnormal posturing
- an inability to get out of bed without help
- more severe speech problems and communication struggles, though the ability to understand what is happening may be retained
- severe psychiatric and behavioral symptoms, though these may not be understood due to communication difficulties
Late-advanced stage Huntington’s
By the final stage of the disease, which typically occurs 11 to 26 years after onset, people with Huntington’s become completely dependent on others for their care. In late stage Huntington’s, people may need to be on a chair or bed most of the time.
Patients with end stage Huntington’s disease will often benefit from round-the-clock specialty care to keep them comfortable.
Average Huntington’s disease life expectancy is in the mid-50s.
Huntington’s Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related articles
-
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
Discussion
-
 Discussion
Discussion
-
Discussion