
Huntington’s disease diagnosis
Last updated Aug. 14, 2025, by Marisa Wexler, MS
 Fact-checked by Marta Figueiredo, PhD
Fact-checked by Marta Figueiredo, PhD
The diagnosis of Huntington’s disease, a progressive disorder that causes problems with movement, cognitive function, and behavior, involves genetic testing to identify a disease-causing mutation paired with clinical assessments to evaluate symptoms. Several screening tools aid in this process.
Median Huntington’s life expectancy after diagnosis is about 15 years. Early Huntington’s disease diagnosis and screening can help people affected by the neurodegenerative condition to plan for the future so that they can maintain the best possible quality of life.
The importance of an early diagnosis
There are no treatments that can slow or stop Huntington’s progression, but getting a prompt diagnosis is vital for allowing patients to explore their options moving forward. Some benefits may be:
- access to medications that can ease symptoms sooner
- faster access to supportive care, including nondrug treatments
- better planning for the future, together with their families
If a person is suspected of having Huntington’s due to a family history of the disease, confirming the diagnosis is generally straightforward. Those without such a family history may face more obstacles in the diagnostic journey, such as needing to rule out other conditions that can cause similar symptoms before moving to genetic testing.
Screening tools for Huntington’s
Huntington’s disease cause is a mutation in the HTT gene that’s characterized by an excessive number of CAG repeats — a sequence of three building blocks of DNA, namely, cytosine (C), adenine (A), and guanine (G). Screening for this gene mutation is the most definitive Huntington’s disease diagnosis test.
Genetic testing for Huntington’s disease looks at the number of CAG repeats in the HTT gene. This is done on a sample of blood or other cells from the body. The results of such testing can determine a person’s likelihood of developing Huntington’s and passing it to their children:
- Up to 26 CAG repeats means no risk of developing Huntington’s.
- Having 27 to 35 repeats indicates that the individual will not develop Huntington’s. However, because repeats of this length are unstable and can expand when transmitted to offspring, there is an increased risk of having biological children with Huntington’s.
- A total of 36 to 39 repeats is classified as a reduced penetrance mutation, meaning some people will develop Huntington’s but others will not. Due to CAG repeat instability, people carrying these many repeats are more likely to have children with Huntington’s.
- Having 40 or more repeats is classified as a full penetrance mutation, meaning it’s essentially guaranteed that the person will develop Huntington’s. Also, each of the individual’s children has a 50% chance of developing the disease.
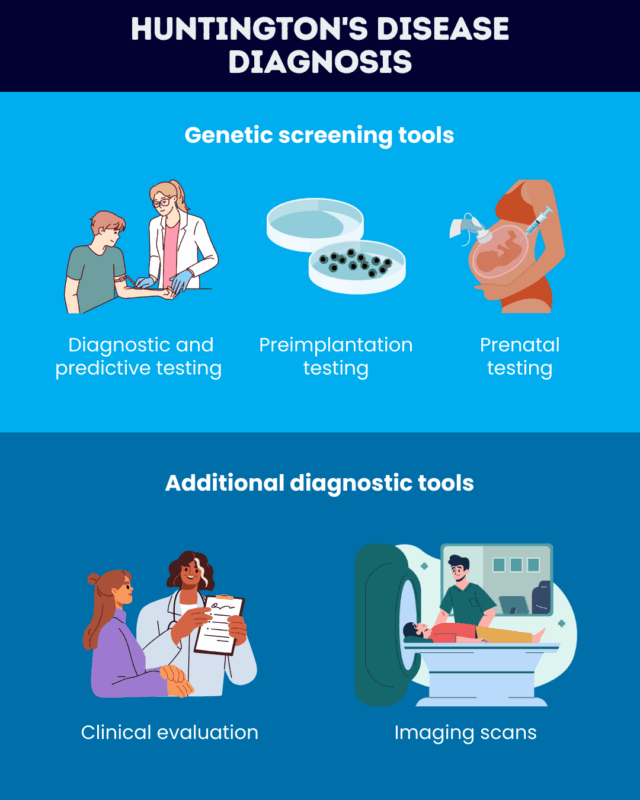
Diagnostic and predictive testing
In people who are experiencing Huntington’s symptoms, genetic testing may be used to confirm the diagnosis, should the person carry 36 or more CAG repeats.
This type of testing can also be used to predict a person’s risk of developing Huntington’s in the future, and may help predict when the disease might occur. Although it’s impossible to predict exactly when symptoms will appear, a general rule is that people with more repeats tend to have an earlier age at onset.
The average age of symptom onset is 35 to 44 years, with the disease being classified as adult-onset Huntington’s, which is typically associated with 40 to 50 CAG repeats. Juvenile Huntington’s disease, marked by symptom onset before age 20, is linked to a higher number of repeats.
Preimplantation genetic testing
For people who are at risk of passing a Huntington’s-causing mutation to their biological children, preimplantation genetic testing (PGT) may be used to reduce this risk. PGT can be done as part of in vitro fertilization, a form of assisted reproductive technology commonly known as IVF.
During IVF, sperm and egg cells are collected and subsequently combined in a laboratory. The resulting embryo is then implanted into the mother’s uterus. Prior to implantation, PGT can be done to see if any of the resulting embryos carry a Huntington’s-causing mutation, allowing only those testing negative to be implanted.
Prenatal testing
Prenatal testing for Huntington’s may be done during pregnancy to determine whether or not the developing fetus carries a disease-causing mutation. It first involves collecting a small sample of fetal cells and DNA, which can be done via several methods:
- Chorionic villus sampling, or CVS, involves the collection of tissue from the placenta using a tube passed through the cervix or a needle inserted into the abdomen. It is usually performed during weeks 10-13 of pregnancy.
- Amniocentesis requires the collection of cells from the fluid that surrounds a developing fetus. It is done by inserting a thin needle into the abdomen, with the procedure usually taking place between 15th and 20th weeks of pregnancy.
Diagnostic tools for Huntington’s
Even if a person is known to carry a Huntington’s-causing mutation, the individual is not said to have the disease until symptoms manifest. Several tools may be used to assess symptoms and track Huntington’s disease progression.
Clinical assessment
During a clinical assessment, an expert will conduct a neurological exam to evaluate potential signs and symptoms of Huntington’s. These may include evaluating a person for motor problems, such as the hallmark involuntary movements known as chorea, as well as cognitive difficulties, and behavioral and mental health issues.
A standardized assessment called the Unified Huntington Disease Rating Scale (UHDRS) is the main tool used to evaluate all these types of Huntington’s symptoms. It assesses various aspects of the disease, including its effects on motor skills, cognitive function, and behaviors, as well as a person’s ability to function independently in day-to-day life.
The UHDRS can be used to help determine when a person carrying a disease-causing mutation has developed Huntington’s. Based on UHDRS scores related to motor function, clinicians can calculate a diagnostic confidence level (DCL) score from zero (no motor signs of Huntington’s) to four (unquestionable signs of Huntington’s).
Manifest Huntington’s may be formally diagnosed if a person has a confirmed disease-causing CAG repeat expansion and other signs, such as:
- motor and cognitive symptoms affecting the individual’s life
- functional decline
- a motor DCL score of three or four
A score of two is considered confirmatory if there are significant cognitive changes and clear evidence of progression.
Following a Huntington’s disease diagnosis, the UHDRS can also be used to track Huntington’s progression over time. This scale includes several validated cognitive tests and functional measures that can be used independently to assess disease progression.
A number of other validated measures of cognitive function and behavior may be used to assess the severity and progression of symptoms.
Imaging scans
As Huntington’s develops and progresses, certain parts of the brain start to shrink. Imaging tools such as an MRI or CT scan may be used to identify and track this brain damage. While these scans aren’t required to diagnose Huntington’s, they may help cement the diagnosis if certain tests are unavailable or inconclusive.
Some people with Huntington’s may have detectable brain changes before symptoms develop, while brain imaging may appear normal in people with early signs of Huntington’s.
Imaging also may be helpful for tracking the disease’s evolution over time.
Benefits and limitations of diagnostic and screening tools
Tests for Huntington’s can provide an estimated risk of Huntington’s, confirm if a person will definitely develop the disease, and determine whether a person is already manifesting the condition.
This, combined with information about a person’s risk of transmitting a disease-causing mutation to any offspring, allows for better planning of a future with or without Huntington’s.
These types of tools also help to get a better understanding of Huntington’s onset and progression, and allow people with confirmed manifest Huntington’s to access care sooner. Individuals with a confirmed diagnosis also may wish to consider participating in Huntington’s research, such as clinical trials.
Still, genetic testing also has some drawbacks. In people with a reduced penetrance mutation, genetic testing cannot determine whether or not they will or won’t develop Huntington’s.
Also, even if a person is found to have a Huntington’s-causing mutation, genetic testing provides limited information about the likely age of onset or disease trajectory. This can be a source of significant emotional stress for patients and their families, particularly because a positive test result confirms an inevitable diagnosis for a disease with no cure or disease-modifying treatments.
As such, people at risk of Huntington’s are often faced with complex choices about whether or not to undergo genetic testing.
Huntington’s Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.