
 Discussion
Discussion
Huntington’s disease overview
Last updated Aug. 26, 2025, by Marisa Wexler, MS
 Fact-checked by Marta Figueiredo, PhD
Fact-checked by Marta Figueiredo, PhD
Huntington’s disease is a genetic disorder that causes progressive damage and death to nerve cells in the brain. It is marked by symptoms including movement disorders, cognitive impairment, and behavioral disturbances.
What is Huntington’s disease?
Huntington’s is a progressive neurodegenerative disorder that especially affects medium spiny neurons, a nerve cell type mostly found in a part of the brain called the striatum that is involved in motor and cognitive functions, as well as emotion. The degeneration of these nerve cells leads to the various neurological symptoms that characterize Huntington’s.
People with Huntington’s disease typically start experiencing symptoms between the ages of 35 and 44, and the average Huntington’s disease life expectancy is in the mid-50s. In about 5% to 10% of cases, symptoms develop before age 20, and the condition is called juvenile Huntington’s disease.
Worldwide, Huntington’s affects approximately 2.7 out of every 100,000 people. Huntington’s disease prevalence is markedly higher in people of Caucasian descent — up to 13 out of every 100,000 people — and lower in people of African or Asian descent.
Causes of Huntington’s disease
All cases of Huntington’s disease are caused by a mutation in the HTT gene. There are no other Huntington’s disease causes.
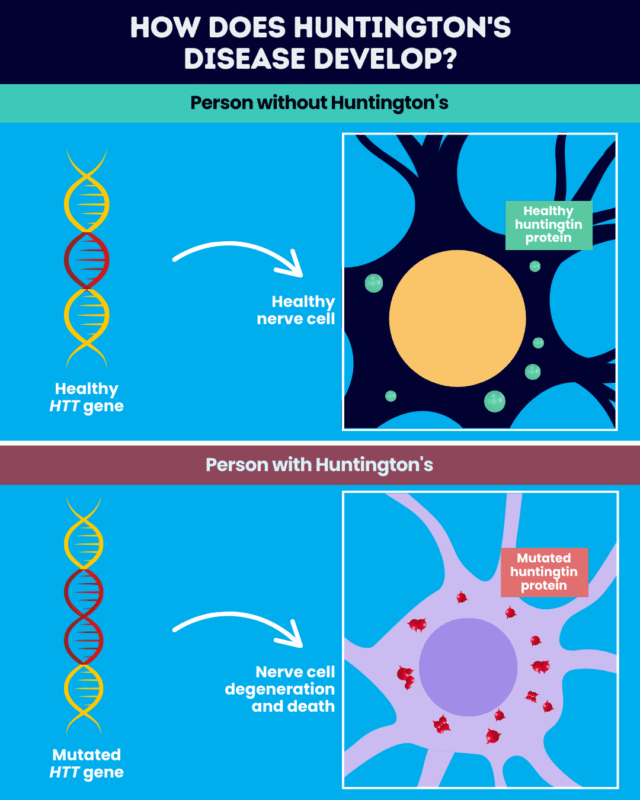
DNA is composed of building blocks called nucleotides. The HTT gene contains a section where a sequence of three nucleotides — cytosine (C), adenine (A), and guanine (G) — is repeated multiple times. In people with Huntington’s, there are many more CAG repeats than normal.
In terms of the number of CAG repeats, Huntington’s disease genetics are classified as:
- Normal: 26 or fewer repeats are considered normal. A person with this number of repeats will not develop Huntington’s.
- Intermediate: 27 to 35 repeats are in the intermediate range. A person carrying this number of repeats will not develop the disease, but is at risk of passing a disease-causing mutation to their biological children.
- Reduced penetrance: 36 to 39 repeats are considered reduced penetrance mutations. People with these many repeats may or may not develop the disease, though the likelihood increases with more repeats.
- Fully penetrant: 40 or more repeats are considered fully penetrant mutations. A person with this number of repeats will almost certainly develop Huntington’s. The age of onset is typically earlier with a higher number of repeats.
Excessive CAG repeats in the HTT gene result in the production of an abnormally long huntingtin protein that is prone to forming toxic aggregates in nerve cells. The toxic effects of mutant huntingtin, along with healthy huntingtin protein dysfunction, are believed to be largely responsible for the nerve damage that drives Huntington’s disease. The precise underlying mechanisms, however, remain a subject of ongoing investigation.
How is Huntington’s inherited?
Everyone inherits two copies of the HTT gene, one from each biological parent. Because Huntington’s is passed down in an autosomal dominant way, a person only needs to inherit one copy of the faulty gene to develop the disease.
This means that if someone with Huntington’s disease has a biological child, there is a 50% chance that the child will inherit a disease-causing mutation and develop the neurodegenerative condition. Genetic testing is available to help inform the future risk of Huntington’s and guide discussions about family planning.
There are no Huntington’s disease risk factors other than a family history of the disorder and the presence of excessive GAG repeats in the HTT gene.
Symptoms of Huntington’s disease
Huntington’s disease symptoms include motor disorders, cognitive problems, and psychiatric issues.
Common motor symptoms of Huntington’s include:
- chorea, or uncontrollable dance-like movements that can affect the arms, legs, or face
- problems with coordination
- rigidity
- balance issues
- slowness of movement
- difficulty speaking and swallowing
People with Huntington’s disease frequently experience cognitive symptoms, including:
- difficulty organizing, planning, or making decisions
- problems with memory and learning
- slow thinking and reaction times
Psychiatric problems that are common in Huntington’s include:
- anxiety
- depression
- mood swings
- apathy
- irritability and aggression
- impulsivity and loss of inhibitions
- psychosis (hallucinations and/or delusions)
Stages of Huntington’s disease
Due to its progressive nature, Huntington’s causes symptoms that begin subtly and worsen over time. While multiple systems are used to describe the stages of Huntington’s disease, a five-stage system is typically used to describe the disease’s progression after symptom onset.
This classification system, based on a person’s level of functional capacity, helps patients and healthcare providers understand disease progression and plan for the future.
- Early Stage: In this initial stage, symptoms such as minor involuntary movements, mood swings, and cognitive difficulties first appear. However, these symptoms do not significantly interfere with daily life, and the person remains fully functional, often able to work and live independently.
- Early-intermediate stage: Symptoms become more prominent and begin to impact day-to-day activities. The person often loses their ability to work and may need help managing finances and household chores. However, they are still able to perform personal care tasks on their own.
- Late-intermediate stage: This stage is marked by a clear decline in functional capacity. Symptoms worsen, requiring the person to rely on others for substantial help with most daily responsibilities. Thinking and memory problems become more severe, and communication can become challenging.
- Early-advanced stage: Symptoms become severe, and the person is no longer able to live independently. They require full assistance with all daily tasks, but may still be able to understand and follow simple instructions.
- Late-advanced stage: This is the final stage, where a person is completely dependent on others for all care. They are often bedridden and may be unable to speak, although they often retain some level of comprehension.
The timeline for progression from early stage Huntington’s to end-stage Huntington’s varies significantly from person to person, but it typically spans about 15 years.
Diagnosing Huntington’s disease
A Huntington’s disease diagnosis is established when a person has a confirmed HTT gene mutation and is experiencing symptoms indicative of Huntington’s. If a person is displaying symptoms but doesn’t know their mutation status, genetic testing is considered 100% accurate in determining whether or not they have Huntington’s.
Other diagnostic tools such as imaging scans may be helpful in monitoring Huntington’s progression, but are generally not needed to confirm the diagnosis.
Huntington’s disease treatment
There is no cure for Huntington’s disease, and no treatment to date has been proven to slow or stop disease progression — though several experimental treatments are currently in development.
In the absence of disease-modifying therapies, Huntington’s disease treatment currently focuses on managing symptoms so that patients can maximize their quality of life.
There are several therapies approved in the U.S. and elsewhere for Huntington’s-associated chorea. Medications such as antidepressants and antipsychotics are often prescribed off-label to manage the psychiatric manifestations of the disease.
People with Huntington’s also commonly benefit from supportive care such as physical therapy, occupational therapy, speech therapy, psychotherapy, and palliative care. Lifestyle modifications, like getting regular exercise, eating a healthy diet, and maintaining consistent routines also can help people with Huntington’s maximize their health and well-being.
Living with Huntington’s disease
Living with Huntington’s disease is challenging, but no one needs to navigate life impacted by the disease on their own. There are support groups and other resources available to provide assistance to people affected by Huntington’s.
Beyond medical treatment, patients and their loved ones can take empowering steps to enhance their quality of life with Huntington’s disease. Some tips for living well with Huntington’s include:
- Prioritizing physical health: Maintaining a balanced diet and engaging in regular exercise.
- Establishing daily routines: Consistent routines can provide a sense of stability and help with memory and organization.
- Staying socially connected: Remaining engaged in social activities and pleasurable hobbies.
- Using medications effectively: Taking all medications exactly as prescribed and communicating openly with healthcare providers about their effects and symptom changes.
- Creating a safe home environment: Modifying the home with safety features and adaptive equipment as needed to prevent falls and ease daily tasks.
- Improving sleep: Practicing good sleep hygiene, as it is essential for both physical and mental well-being.
- Planning for the future: Preparing for the disease’s progression by making explicit plans for future care, legal matters, and financial decisions.
Huntington’s Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related articles
-
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion