
PET Scan Used to Distinguish Juvenile HD From Parkinson’s in Case Study



A teenage boy with a history of parkinsonism was diagnosed with juvenile-onset Huntington’s disease (HD) using high-sensitivity positron emission tomography imaging, known as a PET scan, as described in a recent case study.
The study highlights the importance of using appropriate methods to differentiate between genetic disorders with similar features.
The case report, “Juvenile-onset hypokinetic-rigid Huntington’s disease: a case with dual-tracer positron emission tomography,” was published in the journal Quantitative Imaging in Medicine and Surgery.
Huntington’s, which typically develops in adults ages 30 to 50, is caused by a mutation resulting from an expansion of CAG, a repeated triplet of nucleotides — the building blocks of DNA. CAG repeats expand to as many as 10 times the normal number in Huntington’s, with longer repeats being associated with earlier onset of the disease.
Juvenile-onset Huntington’s, which usually manifests before a patient is 20 years old, is characterized by symptoms atypical of adult-onset HD. These symptoms include gait and speech impairments and seizures, as well as dystonia — involuntary repetitive twisting and sustained muscle contractions — that are more typically observed in Parkinson’s disease.
Due to the similarities in symptoms, juvenile-onset Huntington’s is sometimes misdiagnosed as juvenile-onset Parkinson’s. Genetic testing is required to distinguish between the two diseases.
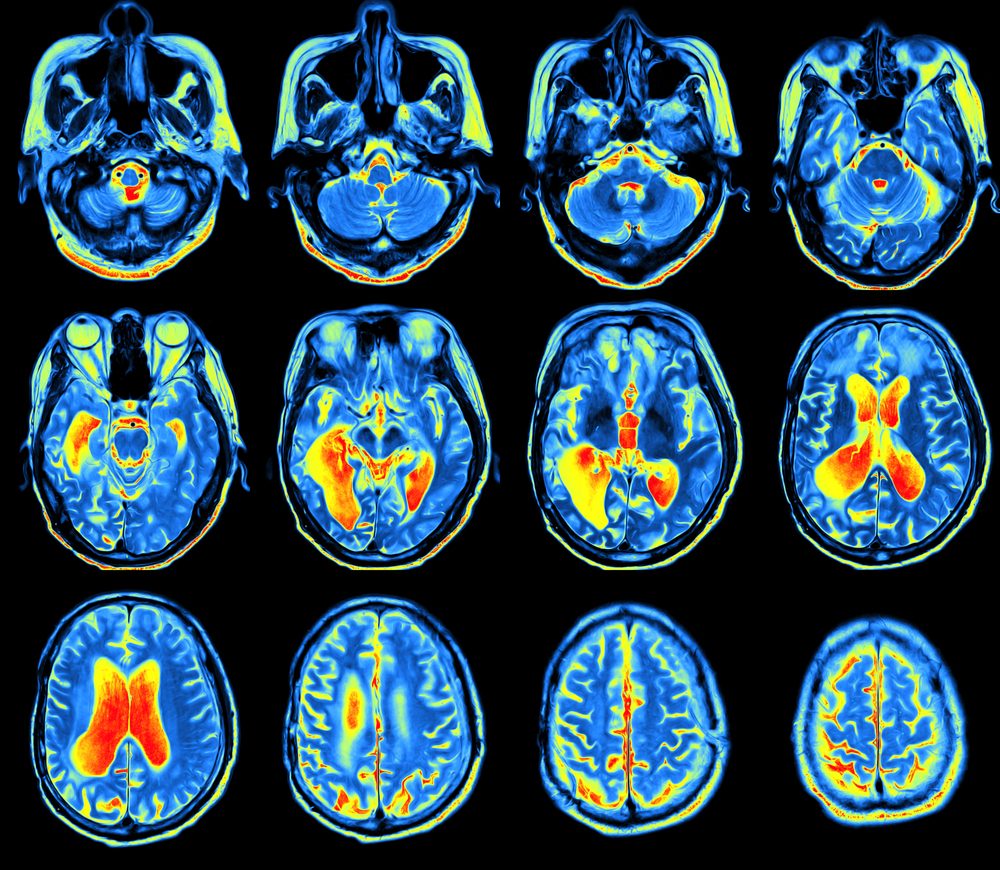
Methods such as magnetic resonance imaging (MRI) and PET scans have been used to diagnose juvenile-onset Huntington’s disease by identifying brain abnormalities.
This case report described the clinicians’ use of dual-tracer PET — which uses two radioactive tracers to visualize brain metabolism — to diagnose juvenile-onset HD in a 16-year-old boy with a two-year history of parkinsonism.
“PET scans have long been used to evaluate patients with [juvenile-onset Huntington’s disease]; however, to our knowledge, this case is the first dual-tracer PET study of [the disease] to be reported,” the researchers said.
The patient displayed slow movements and rigidity that was more prominent on his left side, as well as mild dystonia in his left foot when walking. Moreover, he performed below average on intelligence and memory tests. The patient’s father and uncle experienced involuntary movements starting around the age of 30, as did his grandfather, albeit with an unknown onset age.
The teenager underwent MRI and PET scans that revealed a loss of volume, lesions, and abnormal neurotransmission — the transmission of chemical signals between nerve cells — in the basal nuclei, the area of the brain that controls voluntary movement. These features are present in both Parkinson’s and Huntington’s diseases.
The dual-tracer PET imaging revealed decreased brain glucose consumption in part of the basal nuclei, in contrast to the increased consumption that is characteristic of Parkinson’s. In fact, the observed pattern was more consistent with adult-onset Huntington’s disease.
Based on this and his family history, the patient was diagnosed with juvenile-onset HD, later confirmed by genetic testing.
“In the future, cohort [group] studies focusing on molecular imaging should be carried out to further distinguish between [juvenile-onset Huntington’s disease] and [adult-onset HD], as well as to provide evidence for the neurobiological basis of different clinical presentations,” the researchers concluded.


