Huntington’s May Be Caused by Disrupted Transport of Molecules Within Neurons



Disruption in the trafficking of molecules in and out of the cell’s nucleus may contribute to the pathology of Huntington’s disease, according to results of recent research.
The study “Mutant Huntingtin Disrupts The Nuclear Pore Complex” was published in the journal Neuron.
Huntington’s disease is caused by mutations in the gene encoding the protein huntingtin (HTT), but exactly how these mutations cause deficient neuronal activity is not completely understood.
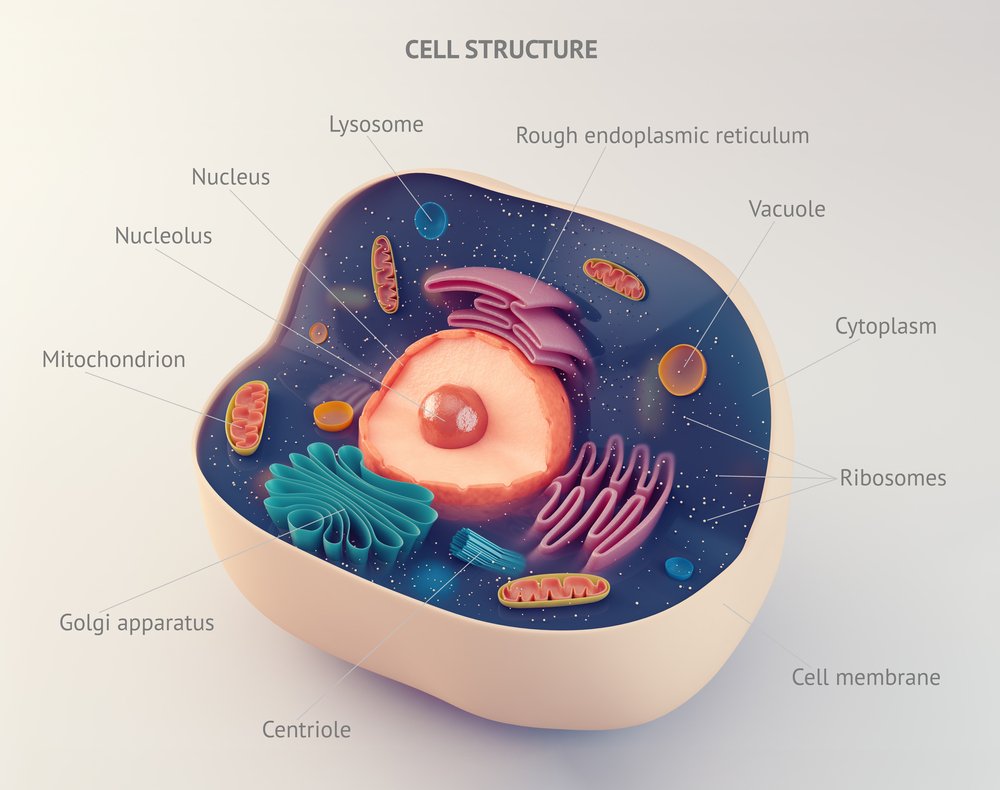
Previous studies have suggested that mutant HTT may interfere with the normal activity of a group of proteins called nuclear pore complexes (NPCs), whose work is to make possible the trafficking of molecules between the cells’ nucleus and cytoplasm (where the rest of the cell’s machinery is located), also known as nucleocytoplasmic transport. One of these proteins is RanGAP1, whose gene has been found to be mutated in Huntington’s patients.
To investigate this hypothesis, researchers used several models of Huntington’s disease, including flies, mice, cultured human neurons and brain samples from patients, to study NPCs’ activity and nucleocytoplasmic transport.
“We’re trying to get at the heart of the mechanism behind neurodegenerative diseases and with this research believe we’ve found one that seems to be commonly disrupted in many of them, suggesting that similar drugs may work for some or all of these disorders,” Jeffrey Rothstein, MD, PhD and the study’s senior researcher, said in a press release.
The team found that the NPC proteins such as RanGAP1 have different locations in disease models and aggregate within cells, which further impairs their role, leading to deficient nucleocytoplasmic transport.
Researchers then tested what happened in mice if they promoted the expression of one additional healthy copy of the RanGAP1 gene together with mutant HTT, compared to the activation of only mutant HTT or a healthy version of HTT.
Results showed that about 17% of neurons with healthy HTT died, compared to 33% of neurons with only the mutant protein. Only 24% of neurons with mutant HTT and RanGAP1 died, suggesting that the presence of healthy RanGAP1 restored part of the nucleocytoplasmic transport.
The team also observed that treating neurons expressing the mutant form of HTT with an experimental drug called KPT-350 improved neuronal survival by preventing a nuclear export protein, called Exportin-1, from transporting molecules out of the nucleus. This result suggests that blocking nuclear export may help neurons survive and revert the effects associated with mutant HTT.
“Our studies show that broken-down components of the nuclear transport machinery lead to traffic jams within brain neurons of essential information and eventually brain cell death,” said Jonathan Grima, the study’s first author. “We believe that the reestablishment of proper cell transport could provide a promising therapeutic target for Huntington’s disease, and potentially other neurodegenerative disorders.”
“We sincerely hope our new findings may help bring us a step closer to treating this and potentially other horrific neurodegenerative disorders,” the researchers added.